Alex Brown
Department of Chemistry University of Alberta
Computational Investigations Complement Experiment for a System of Non-Covalently Bound Asphaltene Model Compounds
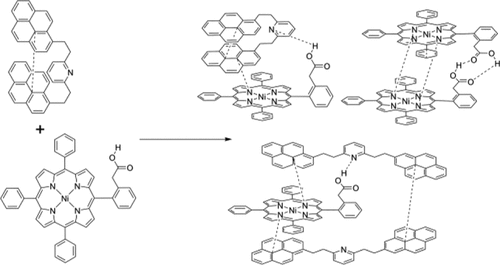
In this paper, the aggregation of asphaltene model compounds has been explored using a combination of an extended tight-binding method (GFN2-xtb) and density functional theory (DFT), in a manner that revisits an experimental study by Schulze, Lechner, Stryker, and Tykwinski (Org. Biomol. Chem. 2015, 13, 6984). The model compounds investigated include a porphyrin with an acidic side chain and a three-island archipelago compound with pyridine as the central island and pyrene for the outer islands. The possible stoichiometries and conformations for complexes were explored and compared to the experimental results. Our computational results show that there are four possible complexes involving these two model compounds with large (K > 1000) equilibrium constants of formation, which will exist in competition with each other. We find that both hydrogen bonding and π–π stacking are important to this aggregation. On the other hand, neither water-mediated aggregation nor coordination to open porphyrin sites was found to be significant in this system, in contrast to some previous suggestions of their importance. The multiple possible stoichiometries of complexes confound some of the analysis done in the experimental paper as Job plots assume that only one complex is present. Gibbs free energies of association were determined for various complexes, with and without microhydration, at the ωB97X-V/def2-QZVPP//ωB97X-D4/def2-SVP level of theory, and using the solvation model based on density (SMD) configured for benzene solvent. We also briefly explore some of the factors influencing the change in NMR chemical shift for select nuclei reported in the experimental paper.